ELSIcon2022 Paper: Population Frequencies of Hereditary Cancer Gene Variants in Diverse Population Groups
ELSIcon2022 • Paper • May 27, 2022
Janis Geary, Kara Hapke, Robert Cook Deegan
Genetic tests to estimate individual risk of various hereditary cancers have proliferated due to improved sequencing technology and in response to interest among clinicians and those at risk. The genes included on germline cancer panels vary substantially from lab to lab. As the market expands, so does the need for data to improve variant interpretations and standards to guide clinical care. Historical underrepresentation of non-Europeans in research and in access to clinical genetic testing can lead to inequities in data required to interpret cancer risks associated with gene variants.
We describe characteristics of hereditary cancer gene variants available through commonly used gene variant databases. We compared hereditary cancer genetic test panels across 17 major companies, and identified 13 genes included in at least one panel from each company. We then downloaded population-based data on the variants of these genes available on the Genome Aggregation Database (gnomAD). We used descriptive statistics to compare population frequency of variants and clinical significance across population groups. We found that pathogenic variant frequency of the 13 genes is not consistent among population groups (e.g.: of the 150 pathogenic/likely pathogenic BRCA1 variants, 86% were found in only 1/8 gnomAD population groups, and 0% in >4), highlighting the need to collect variant data from diverse groups to ensure variants can be interpreted effectively and equitably.
Our analysis provides a snapshot of publicly-available population level data on hereditary cancer gene variants, and demonstrates how crucial it is to ensure non-European populations have data to enable effective variant classifications.
Tags
Videos in Series
-
The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: A Survey on Public Perception toward the Korean National Bio Big Data Project
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Panel: Extending the Belmont Principles: Sovereignty and Solidarity in Genomic Research
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022: Paper Session 12
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Biological sample donation and informed consent for neurobiobanking: Evidence from a community survey in Ghana and Nigeria
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Fresh Air for Genetic Data Sharing
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Public Engagement in Governance of Human Genome Editing in the Post-He Jiankui Era
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: "Extremely slow and capricious:" Genetic Researcher Experiences Using Shared Data Resources
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Consumers’ Perspectives Following Direct-to-Consumer Genetic Testing for Cancer Risk
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022: Flash Session 2
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Incorporating Patient and Relative Preferences in the Design of Traceback Cascade Testing Initiatives
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Flash: Patients’ expectations of benefits from large-panel genomic tumor testing in rural community oncology practices
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022: Moderated Plenary Panel
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Consumer Willingness to Share Personal Digital Information for Health-Related Uses
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Flash: Ensuring equity through insurance coverage for whole exome sequencing and reanalysis
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022: Paper Session 14
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Reproductive Concerns of Disabled Women: Whose Definitions of Disability are We Embracing?
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Flash: Engaging Adolescents in Genomic Decision Making
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Facilitators and barriers to ethical machine learning in healthcare: A qualitative study on developer perspectives of potential harms
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022: Paper Session 13
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Parent-reported barriers and facilitators to accessing care recommended from genomic sequencing in pediatric cancer patients
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Panel: The Ethics of Influence and Choice Architecture in Genomics: Where Have We Been and Where Are We Going?
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Panel: Disability Justice and Precision Medicine Across the Lifespan: Towards a Future of Equity and Access
-
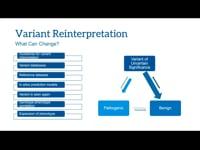
The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Panel: Ethical, Clinical, Legal, and Economic Issues Surrounding Genetic Variant Reinterpretation
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Neglected ELSI in Biobanking: Addressing Issues of Governance, Stewardship, and Equity
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022: Paper Session 17
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Flash: Socioeconomic Status and Interest in Pursuing Genetic Testing for Hereditary Cancer in a US Based Population
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Democratizing Knowledge for Sickle Cell Disease Gene Therapy: A Deliberative Stakeholder Engagement Approach
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Embryoids: What are they, should we be concerned, should they be regulated, and if so, how?
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022: Paper Session 15
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Participation in Sickle Cell Disease Clinical Research: How the Lived Experience Shapes Altruism
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Panel: Engagement with Historically Marginalized Communities: Methods, Positionality and ELSI in Precision Medicine Research
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Why do Primary Care Patients Pursue Genetic Testing as an Elective Clinical Service? Findings from the Sanford Chip Program
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Disrupting the status quo: a new model for benefit sharing in genomics research, a case study from South Africa
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Panel: Towards Justice- Race, ELSI, and Imagined Futures
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Provider Practices in Referring for Germline Genetic Testing for Men with Prostate Cancer at a Safety-Net Hospital
-
The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Networking Session: Salt In My Soul: A Panel Discussion of the Documentary
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Is it just for a screening program to give people all the information they want?
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022: Paper Session 11
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: COVID, Prenatal Genetic Testing, and the Digital Divide: Beyond Technology
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Panel: The Impact of Receipt of Genetic Test Results for Autism
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022: Paper Session 10
-
The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Networking Session: Meet the Experts - Trainee Session
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Cultivating just & equitable genomics research partnerships with community health centers
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Plenary: Troubled Dreams: Genetics, Race, and the Search for Justice in Sickle Cell Disease
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: The Goldilocks Conundrum: Disclosing Discrimination Risks in Informed Consent
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022: Paper Session 9
-
The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Networking Session: Late-Breaking Abstracts, June 2, 2022
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Panel: Voluntary Genetic Testing in the Workplace: An ELSI Analysis
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Genetic data and the collective good: participants as leaders to reconcile individual and public interest
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Translating Genomic Science: How Genetic Counselors Navigate Ethical and Professional Dilemmas in the 21st Century
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Return of Secondary Genomic Findings: Experiences of Sickle Cell Disease Research Participants
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022: Paper Session 5
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Plenary: Ableism, Audism, Ethics, and Genetics: A Just and Equitable Deaf Future?
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Late-Breaking: South Africans’ opinions on human heritable genome editing: Promoting equal access as key to the future
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Child and adolescent psychiatrists’ knowledge, attitudes, and experiences with polygenic risk scores
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Panel: Rare Disease, Equity and Justice: Intersecting Disparities Across the Translational Spectrum
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Navigating community engagement in clinical trial research
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Networking Session: Late-Breaking Abstracts, June 1, 2022
-
The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Why Do Patients Drop Out of Population-Based Screening for Hereditary Cancer Risk?
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022: Paper Session 8
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Mapping Diversity and Equity in Precision Medicine Research: Towards an Ethics of Inclusion
-
The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Networking Session: Ethical Topics Associated with Population Genomic Screening
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022: Paper Session 7
-
The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Mandating Diversity in the Context of Inequity: Re-examining the Research-Care Divide in Precision Medicine Research
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Panel: The Limits of Consent and Patterns of Coercion in U.S. Histories of Eugenic Sterilization
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Panel: What Can Intersectionality Bring to ELSI?: Transforming Theory into Practice
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: How and why do researchers use “Ancestry”?
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Interrogating the Value of Return: Stakeholders’ Perspectives on Return of Results in Biomedical Research
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Sharing Qualitative Research Data: Addressing the Ethical and Logistical Challenges
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Panel: Permeable Membranes: How the blurring of boundaries and merging of silos should reshape ELSI research
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Trainee Networking Session: Working with NHGRI
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Diversity’s Pandemic Distractions
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Exploring Disparities and Equity Across the Newborn Screening System
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Panel: Sharing Data: Speedbumps on the Translational Trail
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022: Paper Session 6
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Late-Breaking: Victims of Eugenic Sterilization in Utah: Demographics and Survivor Estimate
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Clinician experiences and perspectives on the use of polygenic risk scores in child and adolescent psychiatry
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Panel: Development of a Consortium on Best Practices Related to Genomics and Adoption
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Seeking Diverse Inclusion in Bipolar Research: Lessons from a biobank
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: The Individual Genome as a Commons: Getting Genome Conceptualization and Governance Right
-
The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Networking Session: Ethical Aspects of Reproductive Genetic Carrier Screening: Sharing Insights
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Welcome and Keynote: The Fallacy of Biological Race: Systemic Racism in Genomics
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Panel: Developing an Evidence Base for Investigative Genetic Genealogy (IGG) Policy-Making
-

The 5th ELSI Congress - ELSIcon2022 - ELIScon2022 Panel: Polygenic Embryo Screening: Ethical Considerations and Stakeholder Perspectives
-
The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: The Genome and the Biome: Cystic Fibrosis @ Six Feet Apart
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022: Paper Session 4
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Panel: Beyond the Binary: A Panel Discussion About Gender, Genomics, and Justice
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Listening Session: ELSI and the All of Us Research Program
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Flash: Communicating precision medicine research: multidisciplinary teams and diverse communities
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Patients’ and Parents’ Perceived Utility of Genomic Sequencing: Instrument Development and Validation
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022: Paper Session 3
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Attitudes toward and actual uptake of genetic testing among a nationally representative sample of middle-to-older aged U.S. adults
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Cancer's Margins: Exploring Trans* Experiences of Hereditary Cancer Risk
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Genetic testing for hereditary cancer risk in India: A Case Study
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Flash: How Did the Biodeterministic View of Criminality Influence Coercive Sterilization in the State of Iowa from 1934 to 1976?
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022: Paper Session 2
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: “Big bioethics” research: Is the ELSI community ready?
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Networking Session: Late-Breaking Abstracts - May 31, 2022
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: An ELSI-informed strategy to use DNA data for humanitarian family reunifications
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: When State Laws Conflict: Choice of Law Challenges in Multi-Site Research
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022: Paper Session 1
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Flash: What Led to the Abolition of Iowa’s State Eugenics Program in 1977?
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: The Biologization of Morality: Criminal Justice as a Matter of Public Health
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022: Flash Session 1
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Using cost-utility analysis to evaluate the health equity impacts of population genetic screening for BRCA1/BRCA2
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Sociodemographic diversity in clinical genomics research
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Precision medicine for whom? Upstream and downstream exclusion
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Flash: Inequality and Health: Sickle Cell Disease and Food Insecurity during the COVID-19 Pandemic
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Flash: Self-Identified Spirituality and Interest in Genetic Testing for Hereditary Cancer Risk
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Family Secrets Revealed: Experiences and impacts of participating in direct-to-consumer genetic relative-finder services
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Panel: Privacy, Agency, and Relationships in the Genomic Era: Views from the Public
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: The Role of Patient Advocacy Groups Following Prenatal Diagnosis of a Genetic Condition
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Informational Disparities in Electronic Health Records: Implications for Delivery of Hereditary Cancer Genetic Services
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Investigating an Ethos of Duty in Precision Medicine Research Calls for Minority Participation
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Shining a light on overlooked questions in ELSI genomics
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Flash: Genetic Attribution and Felt Stigma in People with Epilepsy
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Race-Based Genetic Testing: Learning from Black Americans to Inform Policy and Practice
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Community Perspectives on Electronic Consent Education for the Retention of Newborn Screening Bloodspots
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: The definition and handling of genomic data in a digitalized society: A Japanese perspective
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Interest in genetic testing offered as part of routine healthcare among an ethnically diverse sample of young women
-
The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Symbolic Legislation and the Regulation of Stroke Biobanking and Genomics Research in Sub-Saharan Africa
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Genetic Options and Constraints: How Genetic Ancestry Tests Change Ethnic Identities
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Flash: Reconstruction of Normality Following the Diagnosis of Ehlers Danlos Syndromes
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Flash: Conceptualization and Measurement of Genetic Literacy
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Flash: Latina women’s knowledge of, preferences for, and experiences with prenatal genetic testing: A scoping review
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: White racial framing and comfort with medical research: a critical analysis
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Systematic review of recommendations on the use of race, ethnicity and ancestry in the context of genetics research
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Panel: How does Genetics and Racial Disparities influence Stillbirth Risk and Reoccurrence?
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Reporting “Clinical Utility” in NICU Sequencing Studies: Systematic Review and Call for Rethinking
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Cystic Fibrosis Communities: Shifting Sites of Prognostic Imagination
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Toward an anthropology of rare disease
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Panel: Out of the tower and onto the ground: engaging communities and scientists in emergent research priorities
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: The 'Lived Experience' of Pediatric Gene Therapy - A Scoping Review
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: How useful is genome sequencing to parents of pediatric cancer patients? Findings from the Texas KidsCanSeq Study
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: ELSI and the New Precision Stewards?
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Resisting an old vice, or how polygenic risk information can do the most good
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Examining Embeddedness: A Call for Principles and Best Practices for Integrating ELSI
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: What I learned about sequencing from my critically ill daughter
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Physician Perspectives on Polygenic Risk Scores
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: The ELSI of Pandemic Biobanking
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: In the Shadows of Virtual Family Trees
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Flash: Revisiting the ethics of secondary findings in diagnostic genome-wide sequencing
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Exploitation in Genetics Research: Vulnerability, Unfairness, and Degradation
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Paper: Whose future should we worry about?
-

The 5th ELSI Congress - ELSIcon2022 - ELSIcon2022 Flash: The Perspective on Healthcare Providers on the Return of Secondary Genetic Findings in Africa